хемофилия
и Мартина Фейхтер, медицински редактор и биологД -р мед. Фабиан Синоватц е на свободна практика в медицинския редакционен екип на
Повече за експертите наМартина Фейхтер учи биология с избираема предметна аптека в Инсбрук и също се потопи в света на лечебните растения. Оттам не стигнаха до други медицински теми, които все още я завладяват и до днес. Тя се обучава като журналист в Академията на Аксел Спрингер в Хамбург и работи за от 2007 г. - първо като редактор, а от 2012 г. като писател на свободна практика.
Повече за експертите на Цялото съдържание на се проверява от медицински журналисти.
Хемофилия (заболяване на кръвта) е нарушение на кръвосъсирването, което обикновено се наследява. На страдащите липсват важни фактори на кръвосъсирването или те са дефектни. В резултат на това хемофилите са склонни към кървене и лесно получават синини. Хемофилията все още не е излекувана. Благодарение на съвременните терапии обаче хемофилиците могат да водят до голяма степен нормален живот. Прочетете всичко, което трябва да знаете за хемофилията тук.
ICD кодове за това заболяване: ICD кодовете са международно признати кодове за медицински диагнози. Те могат да бъдат намерени например в писма на лекар или в удостоверения за неработоспособност. D66D67D68
Кратък преглед
- Какво е хемофилия? Генетично нарушение на кръвосъсирването (коагулопатия). Нарича се още хемофилия.
- Форми на хемофилия: Най-често срещаната е хемофилия А, последвана от хемофилия В. Други форми като хемофилия С, синдром на фон Вилебранд-Юргенс и парахемофилия са по-рядко срещани.
- Причина: Дефицит или дефект на коагулационните фактори (кръвни протеини, необходими за съсирването на кръвта). Той е предимно наследствен и рядко придобит (причинен от спонтанна генна мутация).
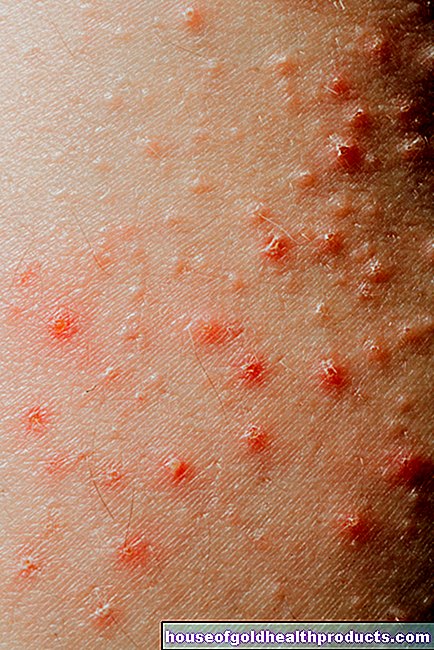
- Симптоми: повишена склонност към кървене, което лесно може да доведе до кървене и синини (хематоми). Кървенето също отнема повече време от нормалното. Колко тежки са симптомите, зависи от тежестта на хемофилията.
- Диагностика: Измерване на различни кръвни параметри (aPTT, бърза стойност, плазмено тромбиново време, време на кървене, брой на тромбоцитите в кръвта), определяне на активността на коагулационните фактори
- Лечение: подмяна на липсващия коагулационен фактор (под формата на факторни концентрати); в някои случаи други лекарства (като десмопресин за лека хемофилия А)
Хемофилия: описание
Хемофилията е вродено нарушение на кръвосъсирването. Засегнатите (хемофилици, "хемофилици") не могат да образуват достатъчно функционални коагулационни фактори. Това са протеини в кръвта, които са необходими за съсирването на кръвта. Поради липсата на коагулационни фактори, кръвни съсиреци и рани не могат лесно да се образуват. Следователно пациентите с хемофилия са склонни към кървене. Те също имат рани, които кървят по -дълго от нормалното. Това може да бъде опасно при определени обстоятелства.
Медицинският термин за нарушение на кръвосъсирването с повишена склонност към кървене е „хеморагична диатеза“. Общото наименование за коагулационно нарушение е "коагулопатия".
Хемофилия: честота
Хемофилията се среща почти изключително при момчета и мъже. Това е сравнително рядко: само около двама от 10 000 мъже имат хемофилия. В Германия са засегнати около 10 000 души. Около 3000 до 5000 от тях имат тежка форма на заболяването.
Хемофилия: форми
Има различни фактори на коагулация. В зависимост от това кой коагулационен фактор е засегнат при хемофилия, медицинските специалисти разграничават различните форми на заболяването.
Хемофилия А.
При хемофилия А има проблеми с коагулационния фактор VIII (антихемофилен глобулин А): Или тялото не може да го произвежда в достатъчно количество, или е дефектно. Около 85 процента от всички хемофилици страдат от хемофилия А. Почти всички от тях са мъже.
Хемофилия В.
При хемофилия В липсва коагулационният фактор IX (антихемофилен глобулин В или коледен фактор). И тук пациентите са предимно мъже. В миналото хемофилия В се е срещала по -често в английското кралско семейство и в руското царско семейство. Затова се нарича още „болест на кралете“.
Други форми на хемофилия
В допълнение към основните форми на хемофилия А и В има и други наследствени нарушения на коагулацията. Един от тях е синдромът на von Willebrand-Jürgens, наричан още синдром на von Willebrand (vWS). Фокусът тук е върху фактора фон Вилебранд: този коагулационен фактор е или твърде малък, или дефектен. Както при хемофилия А и В, това води до повишена склонност към кървене (хеморагична диатеза). В Германия синдромът на von Willebrand-Jürgens се среща при до един процент от населението. За разлика от двете основни форми на хемофилия, това коагулационно разстройство засяга еднакво мъжете и жените.
В редки случаи се появяват и други нарушения на коагулацията, които се основават на липса на коагулационни фактори. Те също имат повишена склонност към кървене (хеморагична диатеза). Те включват:
- Хемофилия C: Дефицит на функционален фактор XI
- Парахемофилия: липса на функциониращ фактор V
- Хипопроконвертинемия: Дефицит на функционален фактор VII
- Дефицит на фактор Стюарт Проуер: Липса на работен фактор X
- Синдром на Хагеман: Дефицит на функциониращ фактор XII
- Дефицит на фибриназа: дефицит на функционален фактор XIII
Хемофилия: симптоми
Симптомите на хемофилия А (дефицит на фактор VIII) и хемофилия В (дефицит на фактор IX) са едни и същи - дори ако при всеки отделен случай са засегнати различни фактори на коагулацията. Като цяло може да се каже: колкото по -малко функционират коагулационните фактори, толкова по -изразена е клиничната картина.
Хемофилия: степени на тежест
Тежестта на хемофилията зависи от това колко е намалена активността на коагулационните фактори в сравнение с тяхната функция при здрави хора. Ако активността на фактора е леко намалена, засегнатите обикновено нямат никакви симптоми. От друга страна, при симптоматичните хемофили активността на фактора е по -намалена, което причинява по -изразени признаци. Има три степени на тежест на хемофилия (А и В):
Лека хемофилия:Факторната активност е 6 до 45 процента от нормалната активност на здрав човек; около 20 % от всички хора с хемофилия имат тази тежест. Засегнатите често не забелязват много от болестта си. Ето защо хемофилията се открива само при много хора в юношеска или зряла възраст, когато операциите или големите наранявания кървят по -дълго от очакваното.
Умерена хемофилия: Факторната активност е 1 до 5 процента от нормалната активност; около 20 % от всички хемофили също са засегнати. Симптомите обикновено стават очевидни през първите няколко години от живота с необичайно продължително кървене и чести синини. Както при лека хемофилия, кървенето обикновено е резултат от наранявания или операция. Спонтанното кървене е рядкост.
Тежка хемофилия: Факторната активност е по -малко от 1 процент от нормалната активност. Това се отнася за около 55 процента от всички хемофилици. Хемофилията обикновено се забелязва още при раждането: премахването на пъпната връв предизвиква масивно кървене. Тежките кръвотечения от носа не са рядкост в ранна детска възраст. В допълнение, дори най -малките наранявания или подутини могат да причинят обилно кървене под кожата (синини). Вътрешното кървене също е по -често, например болезнено кървене в големите стави (като колене, лакти). Обикновено много кръвоизливи нямат видима причина (спонтанно кървене).
Хемофилия: рискове и опасности
Кървенето в ставите (хемартроза) често се случва многократно, особено при тежка хемофилия. В резултат на това засегнатата става може да се деформира, да се износва преждевременно (остеоартрит) и постепенно да се втвърди. Хората с напреднала хемартоза едва могат да движат ръцете и краката си и са зависими от инвалидна количка.
Друга опасност при хемофилия е кървенето в мускулите: може да увреди мускулната тъкан и да доведе до мускулна слабост.
По принцип вътрешно кървене при хемофилия може да възникне във всяка област на органа. Кървенето в мозъка е рядко, но опасно: например, може да наруши способността за мислене и да наруши концентрацията. Тежкият мозъчен кръвоизлив може дори да бъде фатален! Кървенето в корема също може да бъде животозастрашаващо при определени обстоятелства. Силното кървене в орофаринкса може да засегне дихателните пътища.
В допълнение към склонността към кървене (хеморагична диатеза), хемофилия от всякаква степен на тежест може също да доведе до нарушения на зарастването на рани.
Синдром на Von Willebrand-Juergens: Симптоми
Хората със синдром на von Willebrand-Jürgens също имат повишена склонност към кървене. През повечето време има леко кървене под кожата (синини), кървене на венците или продължително кървене след операция, например след екстракция на зъб. Но има и пациенти, които страдат от обилно кървене.
Хемофилия: лечение
Терапията с хемофилия зависи от вида и тежестта на хемофилията. Лекуващият лекар ще създаде подходящ терапевтичен план за всеки пациент. В допълнение към медикаментозното лечение, той може да препоръча и общи мерки. Например, може да се препоръча, особено в случай на тежка хемофилия, да се полагат физически грижи и да се избягват някои (вредни) спортове.
Хемофилия А и В
Днес за лечение на хемофилия се предлагат така наречените концентрати на фактор. Това са концентрати на фактори на кръвосъсирването VIII (за хемофилия А) или IX (за хемофилия В), получени от кръвна плазма или генетично модифицирани. Те трябва да се инжектират във вена (интравенозно). Много страдащи се научават сами да инжектират концентрата на фактора. Това им дава голяма независимост при справяне с болестта.
През 60 -те и 70 -те години на миналия век много хора в Германия се заразяват с хепатит и / или ХИВ от заразени фактори. Това на практика вече не може да се случи днес: кръвната плазма се контролира и третира предварително, преди да се приложи. А с генно инженерните концентрати на фактори обикновено няма риск от инфекция.
В случай на лека и умерена хемофилия, прилагането на концентрат на фактор е необходимо само ако е необходимо (лечение при поискване): Антикоагулантът се прилага, например, в случай на по -тежко кървене или преди планирана операция. Леки наранявания като ожулвания не е необходимо да се лекуват с фактор концентрат. Кървенето обикновено може да бъде спряно чрез лек натиск върху мястото на кървене.
Хората с тежка хемофилия, от друга страна, трябва да си инжектират редовно концентрат на фактор (продължително лечение). Фактор VIII при хемофилия А се прилага два до три пъти седмично. Факторът IX при хемофилия В обикновено трябва да се инжектира само веднъж или два пъти седмично поради по -дългото му време на задържане в кръвта.
Операции и остри наранявания
Преди операциите (дори ако се планира екстракция на зъб), трябва да се проведе подготвителна терапия за всички пациенти с хемофилия. Обикновено за тази цел се прилага фактор концентрат. Това е единственият начин да се предотвратят сериозни усложнения от прекомерна загуба на кръв по време на операцията.
В случай на лека хемофилия А, вместо концентрати на фактор, лекарство, което стабилизира съсирването на кръвта, може да се даде преди планирана операция. Това включва например десмопресин. Това е изкуствено произведен протеин. Той стимулира освобождаването на съхранен фактор VIII от кръвоносните съдове. Лекарството може да се използва само за няколко дни, в противен случай магазините скоро ще бъдат празни.
Съвет: Преди планирана процедура, хората с хемофилия трябва да обсъдят с техния хематолог или център за хемофилия коя превантивна терапия е препоръчителна в техния случай.
В случай на остри наранявания при спешни случаи, освен локални мерки за хемостаза (напр. Превръзки под налягане), е необходимо и прилагането на фактор концентрат.
Фактор концентрат: усложнения
Някои хора образуват антитела (инхибитори) срещу коагулационните фактори във факторния концентрат. Тази така наречена инхибиторна хемофилия е значително по-често срещана при хора с хемофилия А, отколкото при хора с хемофилия В. Инхибиторите инактивират добавения коагулационен фактор. Тогава терапията не е толкова ефективна, колкото се иска. Хемофилия В също заплашва с тежки алергични реакции и други усложнения.
Количеството на инхибиторите в кръвта е дадено в така наречената единица Bethesda (BE). Колкото по -висока е стойността на BE, толкова повече инхибитори има в кръвта на пациента.
При хемофилия А леко натрупване на инхибитори често може да бъде компенсирано чрез увеличаване на дозата на концентрата на фактор. Ако се образуват инхибитори в големи количества, се препоръчва имунопоносима терапия: пациентът получава много високи дози от липсващия коагулационен фактор в комплексната терапевтична схема. Имунната система трябва постепенно да свикне с присъствието си и да спре образуването на инхибитори.
Редкият инхибитор на хемофилия при хемофилия В се лекува по различен начин. Например засегнатите получават лекарства, които засягат имунната система (имуномодулатори).
Болкоуспокояващо
Сериозната хемофилия може да причини силна болка на засегнатите. Например, кървенето в ставите може да бъде много болезнено. Тогава помагат болкоуспокояващи като ибупрофен. За разлика от това, болкоуспокояващото ацетилсалицилова киселина (ASA) е неподходящо за хемофилия: увеличава склонността към кървене още повече (страничен ефект на ASA).
синдром на фон Вилебранд-Юргенс (VWS)
При синдрома на von Willebrand-Jürgens се прави разлика между различни видове, които се лекуват по различен начин: При така наречения тип 1 активната съставка десмопресин се дава, когато е необходимо (преди операция или в случай на остро кървене). Той стимулира освобождаването на съхраняваните коагулационни фактори.
Десмопресин се използва и при тип 2; обаче лекарството не винаги действа тук. След това лекарят предписва вместо това концентрат на фактор (с фактор фон Вилебранд).
Пациентите с VWS тип 3 винаги се лекуват с концентрат на фактор.
Хемофилия: причини и рискови фактори
Хемофилията е вродено генетично заболяване, което обикновено се наследява. По -рядко се случва спонтанно (в резултат на спонтанна промяна на гена = спонтанна мутация).
При хемофилиците генетичната информация, необходима за продуциране на функционален коагулационен фактор, е неправилна: при хемофилия А това е коагулационен фактор VIII, при хемофилия В, фактор IX. Резултатът от грешния строителен план е, че съответният коефициент на коагулация не може да бъде произведен в достатъчно функционално количество. Това нарушава съсирването на кръвта: раните не се затварят толкова бързо, така че кървенето отнема необичайно дълго време. Някои пациенти също имат спонтанно кървене (без видима причина).
Хемофилия А и В: наследяване
Чертежите (гените) за всички части на тялото са върху хромозомите. В ядрото на всяка клетка в тялото има 46 хромозоми, включително две полови хромозоми, които, наред с други неща, определят пола. Жените имат две Х полови хромозоми (XX): Всяка Х хромозома е наследена от майката и една от бащата. Мъжете, от друга страна, имат Y хромозома, наследена от бащата, и X хромозома, наследена от майката (XY).
Гените за коагулационните фактори са на Х хромозомата. При жени, които имат две Х -хромозоми, ако една от тях съдържа дефектен план за коагулационен фактор, това обикновено може да бъде компенсирано от другата Х -хромозома. Следователно те са до голяма степен без симптоми през целия си живот.
Ако такава жена има дете, тя предава една от двете Х хромозоми на потомството. Вероятността това да е копието с дефектен план е 50 процента. Предавайки генетичния дефект, майката става носител (носител) на хемофилията. Ако има син, той ще се роди хемофилик. Дъщерята, от друга страна, обикновено се превръща в потенциален носител на свой ред.
В редки случаи съответният коагулационен фактор не се формира достатъчно дори при женски проводници. След това раните от наранявания или операции могат да кървят дълго време. Още по -рядко е момиче да наследи дефектна Х хромозома от двамата родители. Това е най -вече във връзка със наследственото заболяване синдром на Търнър. Засегнатите момичета показват пълната картина на хемофилия.
Синдром на фон Вилебранд-Юргенс
При синдрома на фон Вилебранд-Юргенс инструкциите за фактора на фон Вилебранд (vWF) показват мутация: коагулационният фактор е твърде малък или е дефектен. Генната мутация може да възникне както при мъже, така и при жени.
Хемофилия: прегледи и диагностика
Ако някой има често спонтанно кървене (като кървене от носа) или много лесно натъртвания, това може да е индикация за хемофилия. Това подозрение е особено очевидно, ако семейството вече е познавало случаи на хемофилия. Засегнатите трябва да изяснят повишената склонност към кървене от лекар. Първата точка за контакт, ако подозирате хемофилия, е вашият семеен лекар:
Лекарят първо ще вземе медицинската история на пациента (анамнеза) в разговор с пациента: Той има подробно описани симптоми, пита за всички основни заболявания и дали има известни случаи на хемофилия в семейството.
Лабораторните тестове са особено важни за изясняване на възможна хемофилия. Лекарят взема кръвна проба от пациента, за да я изследва в лабораторията за различни параметри: При хемофилия, така нареченото aPTT (активирано частично тромбопластиново време) е по-дълго, отколкото при здрави хора. За разлика от това, така наречената бърза стойност (тромбопластиново време, TPZ) и плазменото тромбиново време (PTZ) обикновено са нормални; те се удължават само при тежка хемофилия. Броят на тромбоцитите в кръвта (тромбоцитите) и така нареченото време на кървене също са нормални при хемофилия А и В. При синдрома на фон Вилебранд-Юргенс, от друга страна, времето на кървене се удължава. Времето на кървене е времето, необходимо за спиране на кървенето.
За да може да се определи недвусмислено хемофилия А или В, трябва да се анализира активността на коагулационните фактори (VIII, IX). Специалист по хематология или специализиран медицински център (център за хемофилия) отговаря за това.
Хемофилия: тест при новородени и неродени бебета
Ако едно семейство вече е развило хемофилия, коагулацията на новородените мъже обикновено се проверява веднага след раждането. По този начин хемофилия може да бъде открита на ранен етап. Можете също така да проверите за хемофилия по време на бременност.
Ако една жена подозира, че има генетична предразположеност към хемофилия и следователно е потенциален носител, това може да се изясни с генетичен тест.
Хемофилия: протичане и прогноза на заболяването
Хемофилията все още не е излекувана. Пациентите трябва да се справят с липсата на фактори на съсирването през целия си живот. С помощта на концентрати на фактори обаче те обикновено могат да водят до голяма степен нормален живот.
Ако не се лекува, умерената и тежка хемофилия често водят до сериозни усложнения. Например, кървенето в мускулите може да причини мускулно увреждане. Кръвоизливът в ставите може да доведе до остеоартрит и втвърдяване на ставите. Такива усложнения могат да бъдат избегнати, ако хемофилията бъде открита навреме и се лекува последователно.
Тагове: алтернативна медицина алкохолни наркотици стрес










.jpg)





.jpg)