Фенилкетонурия
Мариан Гросер учи хуманна медицина в Мюнхен. В допълнение, лекарят, който се интересуваше от много неща, се осмели да направи някои вълнуващи заобикалки: изучаване на философия и история на изкуството, работа по радиото и накрая също за Netdoctor.
Повече за експертите на Цялото съдържание на се проверява от медицински журналисти.Фенилкетонурия (PKU) е вродено, наследствено заболяване на протеиновия метаболизъм. Той предотвратява разграждането на аминокиселината фенилаланин. Това се натрупва в организма и нарушава развитието на мозъка на детето. Ако не се лекува, фенилкетонурията води до тежки интелектуални увреждания. С навременната терапия обаче пациентите могат да водят нормален живот. Научете всичко за фенилкетонурията тук!
ICD кодове за това заболяване: ICD кодовете са международно признати кодове за медицински диагнози. Те могат да бъдат намерени например в писма на лекар или в удостоверения за неработоспособност. E70
Фенилкетонурия: описание
Фенилкетонурията (PKU) е наследствено метаболитно заболяване, което съществува от раждането и пречи на разграждането на незаменимата аминокиселина фенилаланин. Аминокиселините са основните градивни елементи на протеините и по този начин жизненоважни метаболитни компоненти. Някои от тях могат да попаднат в тялото само с храната, организмът не може да ги произвежда сам. Такива аминокиселини се наричат незаменими.
Какво се случва с фенилкетонурия?
Обикновено аминокиселините са обект на баланс между усвояване / натрупване и разграждане, така че винаги има толкова налично, колкото е необходимо на тялото. В случай на различни аминокиселини, дефицитът или излишъкът може да причини значителна вреда и да причини различни симптоми.
В така наречената класическа PKU, ефектът на фенилаланин хидроксилаза (PAH) е ограничен или дори напълно отсъства. Поради дефицита на PAH, фенилаланинът все повече се натрупва в организма. Твърде високата концентрация на фенилаланин нарушава значително развитието на мозъка и води до интелектуални затруднения при млади пациенти на ранен етап.
Тъй като нормалното разграждане на фенилаланин не е възможно с болестта, се образуват други продукти на разпадане, така наречените фенил кетони. Те се екскретират с урината и са отговорни за името на болестта.
Атипична фенилкетонурия
Дори при атипични форми на фенилкетонурия, разграждането на фенилаланин се нарушава. Причината обаче не е дефект в ПАХ. Вместо това, функцията на коензим, тетрахидробиоптерин (ВН4), е ограничена. Той косвено участва в разграждането на фенилаланин, тъй като PAH се нуждае от BH4, за да превърне фенилаланин в тирозин.
Тъй като BH4 е важен и за производството на изпращащите вещества допамин и серотонин, атипичната фенилкетонурия с дефицит на BH4 обикновено е по -сложна от класическата форма.
На кого влияе фенилкетонурията?
PKU е едно от най -често срещаните вродени метаболитни заболявания. Смята се, че около едно на 7000 новородени по света ще го развие, без разлика между момичета и момчета. Тъй като това е наследствено заболяване, няколко члена на семейството често са засегнати.
Феникетонурия: симптоми
Първоначално бебетата с фенилкетонурия не показват никакви симптоми на заболяването. Първите проблеми възникват едва на четвъртия до шестия месец от живота, ако болестта все още не е разпозната и лекувана. Преди всичко нарушеното съзряване на мозъка причинява масивни усложнения с течение на времето. Симптомите на нелекуван ПКУ включват:
- силен умствен дефицит. Увреждането на мозъка прогресира през пубертета и след това застоява. След това засегнатите деца обикновено са с тежки умствени увреждания.
- Припадъци (гърчове) (епилепсия). Поради увреждането нервните клетки на мозъка са особено чувствителни и свръхвъзбудими. Резултатът са чести епилептични припадъци.
- двигателни увреждания. Не само мозъчните клетки, но и мускулите на пациента могат да бъдат превъзбудени. Ето защо често става напрегнато (спастичност), което води до различни двигателни нарушения.
- Нарушения на поведението. Някои деца с фенилкетонурия са хиперактивни и необичайно агресивни, а изблиците на гняв също са по -чести.
- малка глава (микроцефалия).Тъй като мозъкът на пациента не се развива правилно, растежът на главата също изостава. Малката обиколка на главата в сравнение с връстниците им е особено забележима при по -големите деца.
- забележима миризма. PKU произвежда определени продукти на разграждане на фенилаланин, които миришат подобно на екскременти от мишки. Тези вещества се екскретират главно с урината, но също и частично през кожата.
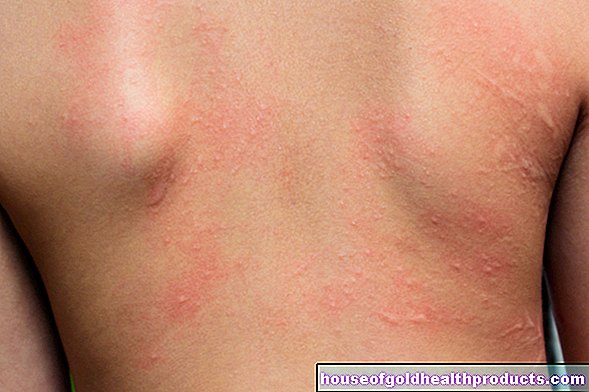
- кожни промени, подобни на екзема
Тъй като производството на пигмент меланин също е нарушено при фенилкетонурия, много страдащи имат много светла, чувствителна към слънцето кожа и бяла руса коса. Ирисът на очите също е светлосин до прозрачен и позволява на червеникавото очно дъно да просветва.
Симптомите на PKU варират по тежест от човек на човек. Основната причина за това е, че активността на фенилаланин хидроксилаза (PAH) е ограничена по различен начин при всеки пациент. Някои все още имат определена остатъчна активност, така че по -малко фенилаланин се натрупва в организма. Други не показват никаква ензимна активност - болестта прогресира съответно по -бързо и по -сериозно.
Фенилкетонурия: причини и рискови фактори
Фенилкетонурията е наследствено заболяване. Понастоящем са известни множество генетични мутации, които водят до дефект в ПАХ. Видът на мутацията определя степента, в която разграждането на фенилаланин е ограничено.
PKU се наследява рецесивно, което означава, че човек може да бъде носител на променен ген, без да развива болестта. По същия начин хората с фенилкетонурия могат да раждат здрави деца.
Само ако и двамата родители имат мутации в генетичния си състав, има известна вероятност тяхното потомство да развие фенилкетонурия. Ако родителите са не само носители на гените, но и сами имат PKU, всички деца заедно също ще го развият.
Фенилкетонурия: прегледи и диагностика
Тъй като сериозните последици от фенилкетонурията могат да бъдат предотвратени чрез своевременно започване на лечение, особено важно е да се открие болестта възможно най -рано. В Германия децата се изследват за различни вродени заболявания, включително ПКУ, като част от общ преглед (скрининг на новородени) на третия ден след раждането.
Тандемна масспектрометрия
Много вродени метаболитни нарушения сега се диагностицират с помощта на така наречената тандемна масспектрометрия. Тя позволява бързо и лесно да се изследва кръвта на новороденото. В допълнение към фенилкетонурията, лекарите могат да открият повече от 20 други заболявания в рамките на няколко минути.
Guthrie тест
Тестът Guthrie, кръстен на своя изобретател, също дава възможност да се диагностицира PKU. За да направите това, малко количество кръв се взема от петата на детето и се нанася върху парче филтърна хартия. След това в лабораторията можете да определите дали концентрацията на фенилаланин е повишена.
Тестът на Guthrie е въведен през 60 -те години на миналия век и отдавна е стандартният метод за диагностициране на фенилкетонурия. Той обаче има недостатъци в сравнение с тандемната масспектрометрия. Тестът Guthrie дава резултат само след пет дни, който също е склонен към грешки. Например фактори като диетата на детето или възможна антибиотична терапия фалшифицират резултатите. В някои страни тестът Guthrie все още се използва, в Германия той обикновено вече не се използва.
Други тестове
Ако скринингът на новороденото разкрие съмнение за фенилкетонурия, следва допълнителен преглед за потвърждение. Това може да се използва и за определяне на точната концентрация на фенилаланин в кръвта.
И накрая, важно е да се разграничи дали е типичен (класически) или нетипичен ПКУ. Предлагат се и специални тестове за това, като стрес тест на тетрахидробиоптерин. Разграничението е важно, тъй като атипичната фенилкетонурия се лекува по различен начин от класическата форма.
Изследване на околоплодни води
Фенилкетонурията вече може да бъде диагностицирана по време на бременност (пренатална диагностика). За да направите това, от амниотичната торбичка на майката се взема малко количество околоплодна течност и се изследват съдържащите се в нея клетки на нероденото дете. Всички генетични дефекти, които причиняват PKU, могат да бъдат идентифицирани по този начин.
При скрининга на новородени обаче фенилкетонурията обикновено се открива достатъчно рано, за да бъде лекувана. Тъй като тестът за околоплодна течност винаги е свързан с определен риск, използването му за диагностициране на ПКУ обикновено не е полезно.
Фенилкетонурия: Лечение
Има само един начин да се противодейства на излишния фенилаланин в ПКУ: Засегнатите деца трябва да спазват специална диета, за да приемат възможно най -малко фенилаланин с храната си. Те също се нуждаят от определени хранителни добавки, които да заменят вещества, които биха се образували от фенилаланин при здрави деца.
Терапията трябва да започне преди появата на първите симптоми на нарушение в развитието, т.е.в рамките на първите два месеца от живота. Вече настъпилите мозъчни увреждания не могат да бъдат отменени.
Хранителна терапия с PKU диета
Всички естествени протеини се състоят от около пет процента от аминокиселината фенилаланин. Повечето храни съдържат много повече от него, отколкото тялото се нуждае. Това не е проблем за здравия човек, тъй като той разгражда и отделя излишните количества фенилаланин.
Пациентите с фенилкетонурия, от друга страна, трябва да се справят в по -голямата си част без естествени протеини. Промишлено произведените специални продукти заместват липсващите хранителни компоненти. От една страна, човек иска да предотврати излишък от фенилаланин, от друга страна, пациентът трябва да бъде снабден с вещества, които иначе биха липсвали, като тирозин, който се образува от фенилаланин.
Целта на диетата PKU обаче не е да спре напълно усвояването на фенилаланин. Тъй като организмът се нуждае от определено количество аминокиселина за важни метаболитни процеси. Постигането на правилната концентрация представлява голямо предизвикателство за лекарите и пациентите и изисква много дисциплина.
Следователно диетата трябва да се спазва цял живот, но особено строго до шестгодишна възраст. Тъй като мозъкът се развива много силно до тази възраст и затова е особено податлив на увреждане. В зряла възраст високите концентрации на фенилаланин не причиняват увреждане на мозъка, както при децата. Те обаче могат да бъдат спусък за други неврологични оплаквания като лоша концентрация или забавени реакции.
Диетичното лечение на фенилкетонурия трябва да започне в специализиран център за метаболитни заболявания. Защото не е възможно във всяка клиника да инструктира родителите за диетата. Това се прави най -добре с помощта на диетично консултиране, което показва как да се придържате към диетата и редовно да наблюдавате нивата на фенилаланин в кръвта.
Лечение на атипична фенилкетонурия
При атипични форми на PKU липсващият коензим BH4 и някои вещества -посланици като допамин и серотонин са изкуствено заменени. В някои случаи пациентът също трябва да е на диета с ниско съдържание на фенилаланин.
Фенилкетонурия по време на бременност
Ако сте бременна и сами имате фенилкетонурия, трябва да имате предвид следното:
- Придържайте се към вашата диета особено строго!
- Проверявайте често кръвните си стойности на фенилаланин в кръвта. Тъй като концентрациите на фенилаланин при нероденото дете са около два пъти по -високи от тези при бременната жена. В допълнение към мозъка, те също могат да увредят сърцето и очите на нероденото дете. Малформациите на скелета на детето също могат да възникнат от високи нива на фенилаланин по време на бременност.
- За да се изключи мозъчното увреждане на детето в ранна бременност, жените с фенилкетонурия трябва внимателно да планират бременността и да вземат предпазни мерки от самото начало.
- При бременни жени с фенилкетонурия диета, която не се спазва стриктно, може да доведе до спонтанен аборт (спонтанен аборт).
- Във всеки случай бременните жени с фенилкетонурия трябва да потърсят съвет и напътствия от лекар.
Фенилкетонурия: протичане и прогноза на заболяването
Ако фенилкетонурията се диагностицира рано, ако е възможно при новороденото, и ако се спазва специалната диета за ПКУ, прогнозата обикновено е добра. Децата се развиват духовно и имат средна продължителност на живота.
Ако не се лекува обаче, увреждането на мозъка води до тежки нарушения на психичното развитие, които не могат да бъдат коригирани по -късно. Засегнатите имат нормална продължителност на живота, но техният коефициент на интелигентност почти винаги е много далеч от нормата.
Рядката атипична форма на фенилкетонурия, при която е налице дефицит на ВН4, е специален случай. Този вариант на фенилкетонурия може да доведе до прогресивно неврологично увреждане с тежки спазми въпреки диетата.
Тагове: ваксинации алкохол зъби